Main Areas of Interest
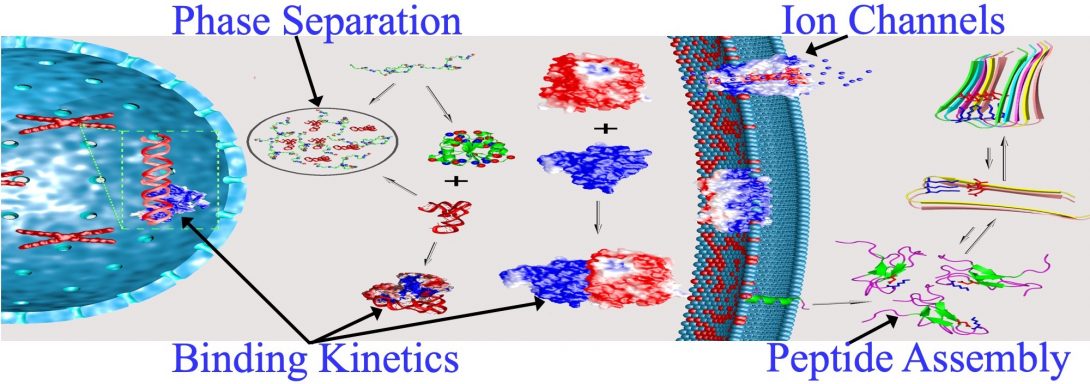
- Liquid-liquid phase separation
- Binding kinetics of intrinsically disordered proteins
- Functional mechanisms of ion channels and other membrane proteins
- Self-assembly of peptides
Liquid-Liquid Phase Separation
The assembly of membraneless organelles via liquid-liquid phase separation is now known to be crucial to many cellular functions. Yet the physical basis of these assembly processes and their delicate regulation is still poorly understood, and hence is the focus of our investigations.
Phase separation occurs because low free energy can be achieved by either decreasing enthalpy or increasing entropy, leading to two phases: the protein-rich droplet phase where protein molecules interact favorably with each other (hence low enthalpy), and the protein-poor bulk phase (with high entropy).
Using simple theoretical models, with structured proteins represented by square-well spherical particles and disordered proteins as chains of square-well beads, we have explained why disordered proteins, in contrast to structured proteins, can readily undergo phase separation (TiBS, 2018).
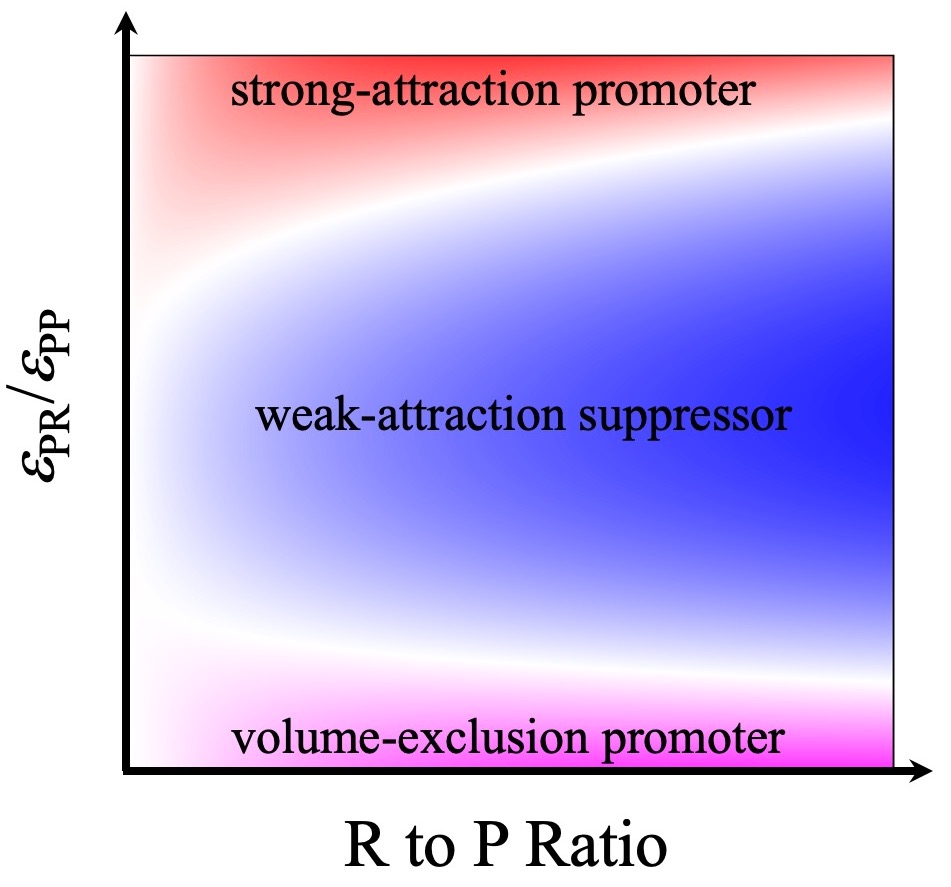
In a 2018 Scientific Reports paper, based on calculations of phase boundaries via Monte Carlo simulations of patchy particles, we identified two important determinants for the effects of macromolecular regulators on the phase boundaries of droplet-forming proteins (P). The first is the strength of attraction (εPR) between P and R; the second is the relative amounts of R and P.
These observations led us to further propose that macromolecular regulators fall into three archetypical classes. Volume-exclusion regulators have only steric repulsion with P; they take up volume in the bulk phase, thereby displacing P into the droplet phase and promoting phase separation. Weak-attraction regulators partition modestly into the droplet phase; once there they replace stronger P-P attraction by weaker P-R attraction, hence suppressing phase separation. Strong-attraction regulators at low concentrations do the opposite. However, at high concentrations, R-R repulsion inside droplets comes into play and thereby strong-attraction regulators switch into suppressors of phase separation. We have now tested these predictions experimentally, marking a crucial step toward establishing a unified understanding of macromolecular regulation of liquid-liquid phase separation (PNAS, 2019).
Several directions are being pursued currently. First, when regulators from different classes are present simultaneously, we have observed nonadditive (“emergent”) effects on phase boundaries. These results are of broad interest because membraneless organelles typically contain dozens to hundreds of macromolecular components; nonadditive effects enable delicate control of the assembly and disassembly of membranelss organelles. Second, macromolecular regulators not only affect phase boundaries but also material properties of the droplet phase; the material properties can have direct implications for cellular functions. Third, in addition to droplets, proteins, with increasing attraction toward each other, can also form gels and aggregates. We are investigating the competition between different condensate phases.
Binding Kinetics of Intrinsically Disordered Proteins
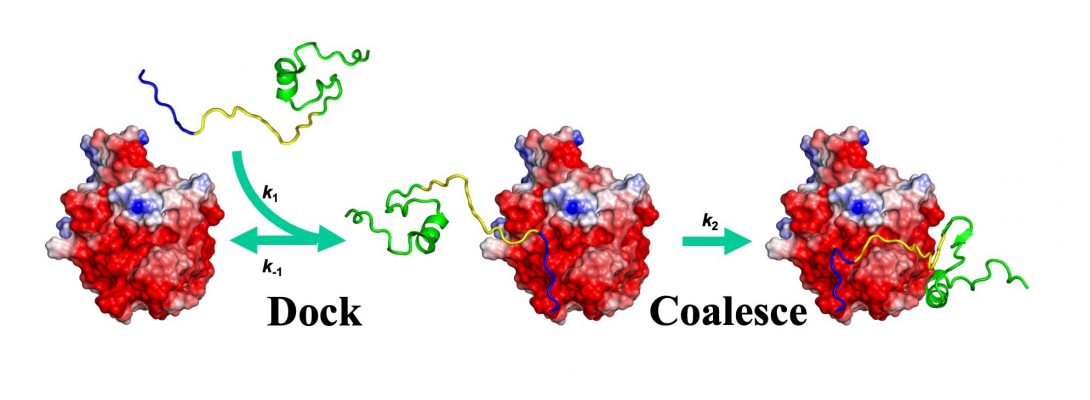
Since 1993, our theoretical and computational studies have contributed to a comprehensive physical understanding on the determinants for the magnitudes of binding rate constants and the determinants for binding pathways (Annu Rev Biophys, 2017). A milestone was the development of TransComp, an automated method for computing protein-protein binding rate constants (Structure, 2011). In this work, we also proposed that the binding of intrinsically disordered proteins to structured targets follows a dock-and-coalesce mechanism. TransComp allows us to model the docking step, but the coalescing step, where conformational changes play a central role, poses a significant challenge to modeling and is a prime target of our ongoing effort.
Fortunately, we can experimentally interrogate the coalescing step. By varying solvent conditions to affect the docking and coalescing steps differentially in stopped-flow kinetic measurements, we have been able to dissect these two steps in the binding of WASP to Cdc42 (FEBS J, 2017).
Potentially, multiple dock-and-coalesce pathways may exist, each initiated by a different docking segment of the disordered protein. One pathway may dominate, as found for the WASP-Cdc42 binding. Here the dominant pathway starts with the electrostatically-accelerated docking of the N-terminal basic region (BR) of WASP, followed by coalescence of the middle CRIB motif and the C-terminal subdomain (Csub). Recently, we have succeeded in altering the dominant pathway by introducing mutations. The mutations involved neutralizing BR cationic residues, to slow down the BR-initiated pathway, and reversing Cdc42 anionic residues around the Csub binding site, to accelerate the Csub-initiated pathway (Sci Rep, 2019).
Current work aims to develop single-molecule probes, based on optical tweezers, to capture intermediates along the dock-and-coalesce pathway.
Ion Channels and Other Membrane Proteins
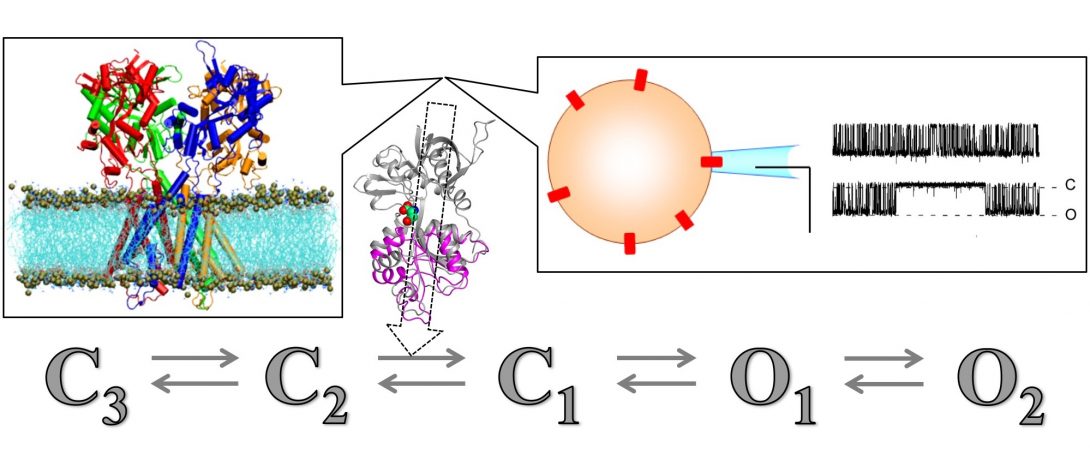
Using data from solid-state NMR and other structural techniques, we are calculating structures of ion channels and other membrane proteins inside membranes. These structures capture but a few snapshots of the proteins during their functional processes. Many other states can only be indirectly probed by experiments, notably ion current recordings through a single channel. We are using computation, in particular molecular dynamics simulations, to provide structural models for states not accessible by experiments and to define pathways for the transitions between states.
Our computational studies have produced mechanistic models for the M2 proton channel of the flu virus (Science, 2010) and for ionotropic glutamate receptors (Nat Commun, 2011; Acc Chem Res, 2017). The latter are ligand-gated ion channels that form the basis of learning and memory. Our studies have profound implication for understanding missense mutations associated with neurological disorders and for developing precision medicine (Trends Neurosci, 2017; Nat Commun, 2018).
Many transmembrane proteins have disordered cytoplasmic regions that associate with membranes. We are using molecular dynamics simulations to elucidate how membrane association contributes to functional mechanisms of these proteins.
Peptide Self-Assembly
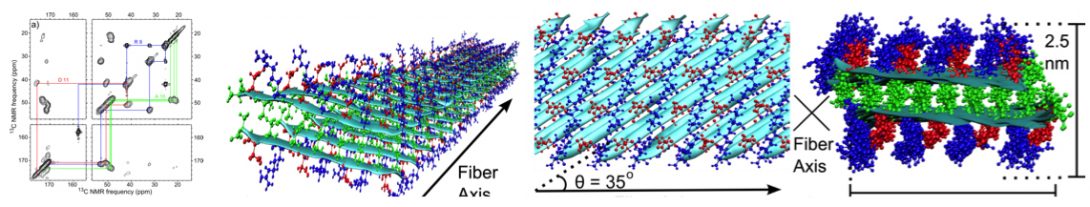
Based on structural restraints from solid-state NMR, we have modeled the molecular structures of nanofibers formed by short peptides (ACS Nano, 2013; Biophys J, 2013).
Extending this integrated approach, we are working on the structural determination of a 150-kD oligomer formed by the Aβ-42 peptide. Through collaboration, we have added cryo-EM to this effort. We are also investigating the assembly pathways of this oligomer.